- Biopython - Home
- Biopython - Introduction
- Biopython - Installation
- Creating Simple Application
- Biopython - Sequence
- Advanced Sequence Operations
- Sequence I/O Operations
- Biopython - Sequence Alignments
- Biopython - Overview of BLAST
- Biopython - Entrez Database
- Biopython - PDB Module
- Biopython - Motif Objects
- Biopython - BioSQL Module
- Biopython - Population Genetics
- Biopython - Genome Analysis
- Biopython - Phenotype Microarray
- Biopython - Plotting
- Biopython - Cluster Analysis
- Biopython - Machine Learning
- Biopython - Testing Techniques
Biopython Resources
Biopython - Plotting
This chapter explains about how to plot sequences. Before moving to this topic, let us understand the basics of plotting.
Plotting
Matplotlib is a Python plotting library which produces quality figures in a variety of formats. We can create different types of plots like line chart, histograms, bar chart, pie chart, scatter chart, etc.
pyLab is a module that belongs to the matplotlib which combines the numerical module numpy with the graphical plotting module pyplot.Biopython uses pylab module for plotting sequences. To do this, we need to import the below code −
>>>import pylab
Before importing, we need to install the matplotlib package using pip command with the command given below −
(myenv) D:\biopython\myenv>pip3 install matplotlib
Sample Input File - plot.fasta
Create a sample file named plot.fasta in your Biopython directory and add the following changes −
>gi|2765658|emb|Z78533.1|CIZ78533 C.irapeanum 5.8S rRNA gene and ITS1 and ITS2 DNA CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGGAATAAACGATCGAGTG AATCCGGAGGACCGGTGTACTCAGCTCACCGGGGGCATTGCTCCCGTGGTGACCCTGATTTGTTGTTGGG CCGCCTCGGGAGCGTCCATGGCGGGTTTGAACCTCTAGCCCGGCGCAGTTTGGGCGCCAAGCCATATGAA AGCATCACCGGCGAATGGCATTGTCTTCCCCAAAACCCGGAGCGGCGGCGTGCTGTCGCGTGCCCAATGA ATTTTGATGACTCTCGCAAACGGGAATCTTGGCTCTTTGCATCGGATGGAAGGACGCAGCGAAATGCGAT AAGTGGTGTGAATTGCAAGATCCCGTGAACCATCGAGTCTTTTGAACGCAAGTTGCGCCCGAGGCCATCA GGCTAAGGGCACGCCTGCTTGGGCGTCGCGCTTCGTCTCTCTCCTGCCAATGCTTGCCCGGCATACAGCC AGGCCGGCGTGGTGCGGATGTGAAAGATTGGCCCCTTGTGCCTAGGTGCGGCGGGTCCAAGAGCTGGTGT TTTGATGGCCCGGAACCCGGCAAGAGGTGGACGGATGCTGGCAGCAGCTGCCGTGCGAATCCCCCATGTT GTCGTGCTTGTCGGACAGGCAGGAGAACCCTTCCGAACCCCAATGGAGGGCGGTTGACCGCCATTCGGAT GTGACCCCAGGTCAGGCGGGGGCACCCGCTGAGTTTACGC >gi|2765657|emb|Z78532.1|CCZ78532 C.californicum 5.8S rRNA gene and ITS1 and ITS2 DNA CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAGAATATATGATCGAGTG AATCTGGAGGACCTGTGGTAACTCAGCTCGTCGTGGCACTGCTTTTGTCGTGACCCTGCTTTGTTGTTGG GCCTCCTCAAGAGCTTTCATGGCAGGTTTGAACTTTAGTACGGTGCAGTTTGCGCCAAGTCATATAAAGC ATCACTGATGAATGACATTATTGTCAGAAAAAATCAGAGGGGCAGTATGCTACTGAGCATGCCAGTGAAT TTTTATGACTCTCGCAACGGATATCTTGGCTCTAACATCGATGAAGAACGCAGCTAAATGCGATAAGTGG TGTGAATTGCAGAATCCCGTGAACCATCGAGTCTTTGAACGCAAGTTGCGCTCGAGGCCATCAGGCTAAG GGCACGCCTGCCTGGGCGTCGTGTGTTGCGTCTCTCCTACCAATGCTTGCTTGGCATATCGCTAAGCTGG CATTATACGGATGTGAATGATTGGCCCCTTGTGCCTAGGTGCGGTGGGTCTAAGGATTGTTGCTTTGATG GGTAGGAATGTGGCACGAGGTGGAGAATGCTAACAGTCATAAGGCTGCTATTTGAATCCCCCATGTTGTT GTATTTTTTCGAACCTACACAAGAACCTAATTGAACCCCAATGGAGCTAAAATAACCATTGGGCAGTTGA TTTCCATTCAGATGCGACCCCAGGTCAGGCGGGGCCACCCGCTGAGTTGAGGC >gi|2765656|emb|Z78531.1|CFZ78531 C.fasciculatum 5.8S rRNA gene and ITS1 and ITS2 DNA CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAGCAGAACATACGATCGAGTG AATCCGGAGGACCCGTGGTTACACGGCTCACCGTGGCTTTGCTCTCGTGGTGAACCCGGTTTGCGACCGG GCCGCCTCGGGAACTTTCATGGCGGGTTTGAACGTCTAGCGCGGCGCAGTTTGCGCCAAGTCATATGGAG CGTCACCGATGGATGGCATTTTTGTCAAGAAAAACTCGGAGGGGCGGCGTCTGTTGCGCGTGCCAATGAA TTTATGACGACTCTCGGCAACGGGATATCTGGCTCTTGCATCGATGAAGAACGCAGCGAAATGCGATAAG TGGTGTGAATTGCAGAATCCCGCGAACCATCGAGTCTTTGAACGCAAGTTGCGCCCGAGGCCATCAGGCT AAGGGCACGCCTGCCTGGGCGTCGTGTGCTGCGTCTCTCCTGATAATGCTTGATTGGCATGCGGCTAGTC TGTCATTGTGAGGACGTGAAAGATTGGCCCCTTGCGCCTAGGTGCGGCGGGTCTAAGCATCGGTGTTCTG ATGGCCCGGAACTTGGCAGTAGGTGGAGGATGCTGGCAGCCGCAAGGCTGCCGTTCGAATCCCCCGTGTT GTCGTACTCGTCAGGCCTACAGAAGAACCTGTTTGAACCCCCAGTGGACGCAAAACCGCCCTCGGGCGGT GATTTCCATTCAGATGCGACCCCAGTCAGGCGGGCCACCCGTGAGTAA >gi|2765655|emb|Z78530.1|CMZ78530 C.margaritaceum 5.8S rRNA gene and ITS1 and ITS2 DNA CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAAACAACATAATAAACGATTGAGTG AATCTGGAGGACTTGTGGTAATTTGGCTCGCTAGGGATATCCTTTTGTGGTGACCATGATTTGTCATTGG GCCTCATTGAGAGCTTTCATGGCGGGTTTGAACCTCTAGCACGGTCCAGTTTGCACCAAGGTATATAAAG AATCACCGATGAATGACATTATTGCCCCACACAACGTCGGAGGTGTGGTGTGTTAATGTTCATTCCAATG AATTTTGATGACTCTCGGCAGACGGATATCTTGACTCTTGCATCGATGAAGAACGCACCGAAATGTGATA AGTGGTGTGAATTGCAGAATCCCGTGAACCATCGAGTCTTTGAACGCAAGTTGCGCCCGAGGCCATCAGG CTAAGGGCACGCCTGCCTGGGCGTCGTATGTTTTATCTCTCCTTCCAATGCTTGTCCAGCATATAGCTAG GCCATCATTGTGTGGATGTGAAAGATTGGCCCCTTGTGCTTAGGTGCGGTGGGTCTAAGGATATGTGTTT TGATGGTCTGAAACTTGGCAAGAGGTGGAGGATGCTGGCAGCCGCAAGGCTATTGTTTGAATCCCCCATG TTGTCATGTTTGTTGGGCCTATAGAACAACTTGTTTGGACCCTAATTAAGGCAAAACAATCCTTGGGTGG TTGATTTCCAATCAGATGCGACCCCAGTCAGGGGGCCACCCCAT >gi|2765654|emb|Z78529.1|CLZ78529 C.lichiangense 5.8S rRNA gene and ITS1 and ITS2 DNA ACGGCGAGCTGCCGAAGGACATTGTTGAGACAGCAGAATATACGATTGAGTGAATCTGGAGGACTTGTGG TTATTTGGCTCGCTAGGGATTTCCTTTTGTGGTGACCATGATTTGTCATTGGGCCTCATTGAGAGCTTTC ATGGCGGGTTTGAACCTCTAGCACGGTGCAGTTTGCACCAAGGTATATAAAGAATCACCGATGAATGACA TTATTGTCAAAAAAGTCGGAGGTGTGGTGTGTTATTGGTCATGCCAATGAATTGTTGATGACTCTCGCCG AGGGATATCTTGGCTCTTGCATCGATGAAGAATCCCACCGAAATGTGATAAGTGGTGTGAATTGCAGAAT CCCGTGAACCATCGAGTCTTTGAACGCAAGTTGCGCCCGAGGCCATCAGGCTAAGGGCACGCCTGCCTGG GCGTCGTATGTTTTATCTCTCCTTCCAATGCTTGTCCAGCATATAGCTAGGCCATCATTGTGTGGATGTG AAAGATTGGCCCCTTGTGCTTAGGTGCGGTGGGTCTAAGGATATGTGTTTTGATGGTCTGAAACTTGGCA AGAGGTGGAGGATGCTGGCAGCCGCAAGGCTATTGTTTGAATCCCCCATGTTGTCATATTTGTTGGGCCT ATAGAACAACTTGTTTGGACCCTAATTAAGGCAAAACAATCCTTGGGTGGTTGATTTCCAATCAGATGCG ACCCCAGTCAGCGGGCCACCAGCTGAGCTAAAA
Line Plot
Now, let us create a simple line plot for the above fasta file.
Step 1 − Import SeqIO module to read fasta file.
>>> from Bio import SeqIO
Step 2 − Parse the input file.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
>>> len(records)
5
>>> max(records)
753
>>> min(records)
733
Step 3 − Let us import pylab module.
>>> import pylab
Step 4 − Configure the line chart by assigning x and y axis labels.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>
Step 5 − Configure the line chart by setting grid display.
>>> pylab.grid()
Step 6 − Draw simple line chart by calling plot method and supplying records as input.
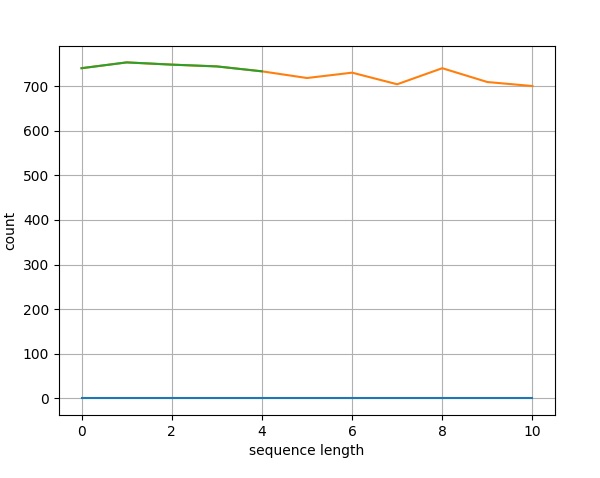
>>> pylab.plot(records) [<matplotlib.lines.Line2D object at 0x000001CDC4BC6D80>]
Step 7 − Finally save the chart using the below command.
>>> pylab.savefig("lines.png")
Result
After executing the above command, you could see the following image saved in your Biopython directory.
Histogram Chart
A histogram is used for continuous data, where the bins represent ranges of data. Drawing histogram is same as line chart except pylab.plot. Instead, call hist method of pylab module with records and some custum value for bins (5). The complete coding is as follows −
Step 1 − Import SeqIO module to read fasta file.
>>> from Bio import SeqIO
Step 2 − Parse the input file.
>>> records = [len(rec) for rec in SeqIO.parse("plot.fasta", "fasta")]
>>> len(records)
5
>>> max(records)
753
>>> min(records)
733
Step 3 − Let us import pylab module.
>>> import pylab
Step 4 − Configure the line chart by assigning x and y axis labels.
>>> pylab.xlabel("sequence length")
Text(0.5, 0, 'sequence length')
>>> pylab.ylabel("count")
Text(0, 0.5, 'count')
>>>
Step 5 − Configure the line chart by setting grid display.
>>> pylab.grid()
Step 6 − Draw simple line chart by calling plot method and supplying records as input.
>>> pylab.hist(records,bins=5) array([1., 1., 1., 1., 1.]), array([733., 737., 741., 745., 749., 753.]), <BarContainer object of 5 artists>) >>>
Step 7 − Finally save the chart using the below command.
>>> pylab.savefig("hist.png")
Result
After executing the above command, you could see the histogram saved in your Biopython directory.
